Overview of Wilson’s Disease:
Wilson’s disease (WD; also known as hepatolenticular degeneration) is an autosomal recessive disorder that causes an excess copper accumulation in the body. It primarily affects the liver and basal ganglia of the brain, but it can also affect other organ systems.
This autosomal recessive disease is caused by a mutation in the Wilson disease protein (ATP7B) gene. For an individual to be affected, a copy of the gene from each parent needs to be inherited. Diagnosis of WD is complex and involves blood tests, urine tests, and a liver biopsy together with the clinical evaluation. Genetic testing may be useful to screen the family members of those affected.
The genetic defect is localized to the long arm of chromosome 13 (13q), which has been shown to alter the copper-transporting ATP gene in the liver. The majority of patients with Wilson’s disease present within the first decade of life with liver dysfunction. The neuropsychiatric features occur in the third/fourth decade of life. Wilson’s disease (WD) is rare but if not recognized in and timely treated, it is fatal.
Epidemiology:
This disease affects 1 in every 30,000 people with a carrier frequency of 1 in every 90. Some populations have a greater incidence of Wilson’s disease due to the increased rate of consanguinous marriages. Both men and women are affected equally. The usual age of presentation is four to 40 years, but this disorder has been detected in children as young as 3 as well as adults as old as 70.
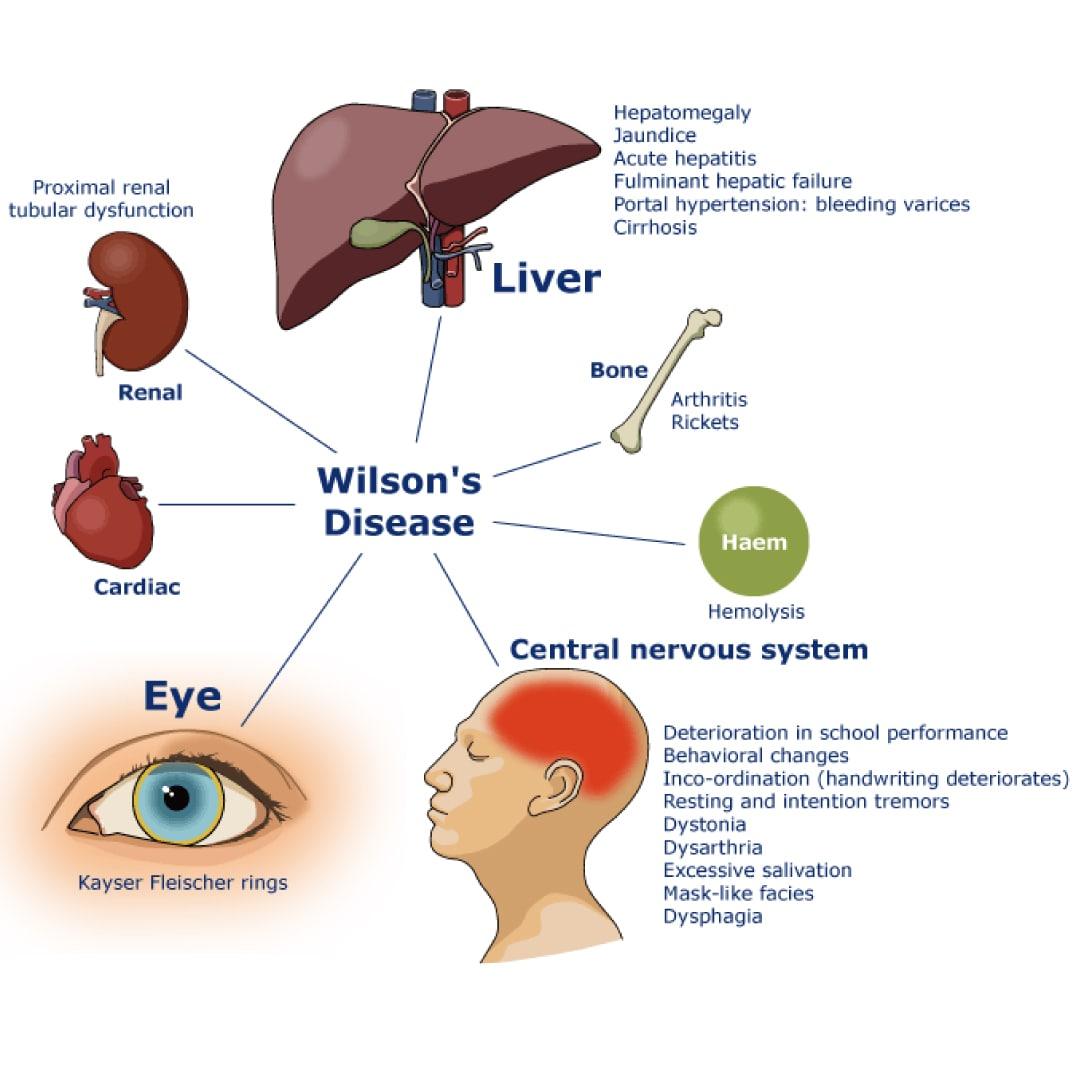
Signs and Symptoms: Signs and symptoms of Wilson’s disease (WD) usually are related to the brain and liver.
Hepatic Symptoms:
- Acute liver failure
- Isolated splenomegaly
- Persistently elevated serum aminotransferase activity (AST, ALT)
- Asymptomatic hepatomegaly
- Fatty liver
- Resembling autoimmune hepatitis
- Acute hepatitis
- Cirrhosis: compensated or decompensated
Neurological Symptoms:
- Insomnia
- Seizures
- Drooling, dysarthria
- Dysphagia
- Pseudobulbar palsy
- Rigid dystonia
- Dysautonomia
- Migraine headaches
- Movement disorders (tremor, involuntary movements)
Psychiatric Symptoms:
- Anxiety
- Depression
- Psychosis
- Neurotic behaviors
- Personality changes
Other Systems:
- Pancreatitis
- Hypoparathyroidism
- Ocular: Kayser-Fleischer rings, sunflower cataracts
- Kidney: Renal abnormalities: aminoaciduria and nephrolithiasis
- Skeletal abnormalities: premature osteoporosis and arthritis
- Cutaneous: lunulae ceruleae
- Cardiomyopathy, dysrhythmias
- Menstrual irregularities; infertility, repeated miscarriages
Treatment of Wilson’s Disease:
Wilson’s disease or WD is very treatable disorder. With proper therapy, disease progress can be stopped and oftentimes signs and symptoms can be improved. The primary goal of treatment is to remove excess accumulated copper and prevent its reaccumulation. Treatment for WD is a lifelong procedure. Patients may become progressively sicker from day to day, so prompt treatment can be critical. Treatment delays may be responsible for causing irreversible damage.
Chelation therapy medicines approved for treating Wilson’s disease (WD) include penicillamine, trientine dihydrochloride, and trientine tetrahydrochloride. These therapeutic drugs act by chelation or binding of copper, causing its increased urinary excretion. The mainstay treatment for WD is copper chelation therapy with penicillamine and trientine. Treatment with Trientine is preferred because of minimal side effects.
Oral zinc also may be useful as it competes for absorption with copper at metallic ion transporter. During pregnancy, d-penicillamine can be used as it does not pose any risk to the fetus. Liver transplantation can be beneficial in improving neurological dysfunction in some individuals not responding adequately to medical therapy.
A low in copper-containing diet/food is recommended with avoidance of nuts, mushrooms, dried fruit, chocolate, liver, and shellfish.
References:
https://www.ncbi.nlm.nih.gov/books/NBK441990/
https://www.aasld.org/practice-guidelines/diagnosis-and-treatment-wilson-disease